白藜芦醇衍生物的发现作为新型LSD1抑制剂:设计、合成及其生物学评价
原文作者:Yi-Chao Zhenga,Yuan-Yuan Guan b, Xiao-Yu Zhai a, Li Na Ding c, Wen-Ping Qin a, Dan-Dan Shen c, Xue-Qi Liu c, Xu-Dong Sun c, Yi-Chao Zheng c, **, Hong-Min Liu c,* 单位:Xinxiang Medical University
摘要:抑制赖氨酸特异性的去甲基化酶1(LSD1)的治疗近来成为治疗癌症和其他疾病的一个强有力方法。我们连续不间断地识别新型小分子LSD1抑制剂并设计合成了一系列白藜芦醇衍生物,而它正是LSD1的有效抑制剂。其中,化合物4e和化合物4m的IC50值分别为121nM和123nM,显示出了最强的LSD1抑制活性。生物化学研究和对接分析表明,化合物4e和4m是可逆的LSD1抑制剂。高含量分析表明,4e和4m对诱导组蛋白H3的二甲基化LYS4的剂量依赖性增加,对MGC-803细胞中LSD1的表达没有影响。此外,4e或4m可以显著增加CD86的mRNA水平,CD86是LSD1活性的细胞生物标志物替代。说明它们可能在MGC-803细胞中表现出LSD1抑制活性。这些发现应该可以发展对这些化合物进行进一步的修饰,以研发具有潜在抗癌活性的更强效的LSD1抑制剂。
关键词:白藜芦醇衍生物; LSD1; 抑制剂合成
- 介绍
组蛋白赖氨酸特异性脱甲基酶1(LSD1)是第一个被鉴定为负责特异性去除单和二甲基化的H3K4和H3K9[1]甲基的脱甲基酶,LSD1还可以脱去许多非组蛋白的甲基,如p53[2],DNA甲基转移酶(DNMTs)[3],E2F转录因子1(E2F1)[4],肌球蛋白磷酸酯酶目标子1(MYPT1)[5]。这种遗传翻译修饰调节许多生物过程,包括细胞分化,基因激活和基因抑制[6]。最近的研究报道,LSD1在多种类型的恶性肿瘤中异常地过度表达,这与肿瘤的发生和发展有关,减少了分化,且引发不良预化[7-15]。LSD1的消除对于急性髓系白血病(AML)[16],乳房[17,18],肺[19],胃[20],直肠癌等[21],可诱导多种表观遗传学沉默肿瘤抑制基因来抑制细胞增殖、迁移和侵袭。据报道当LSD1抑制剂与泛高密度脂蛋白胆固醇抑制剂组合时表现出协同抗癌活性[22-26]。因此,LSD1已成为治疗人类恶性肿瘤的一个有前途的靶点[27-29]。
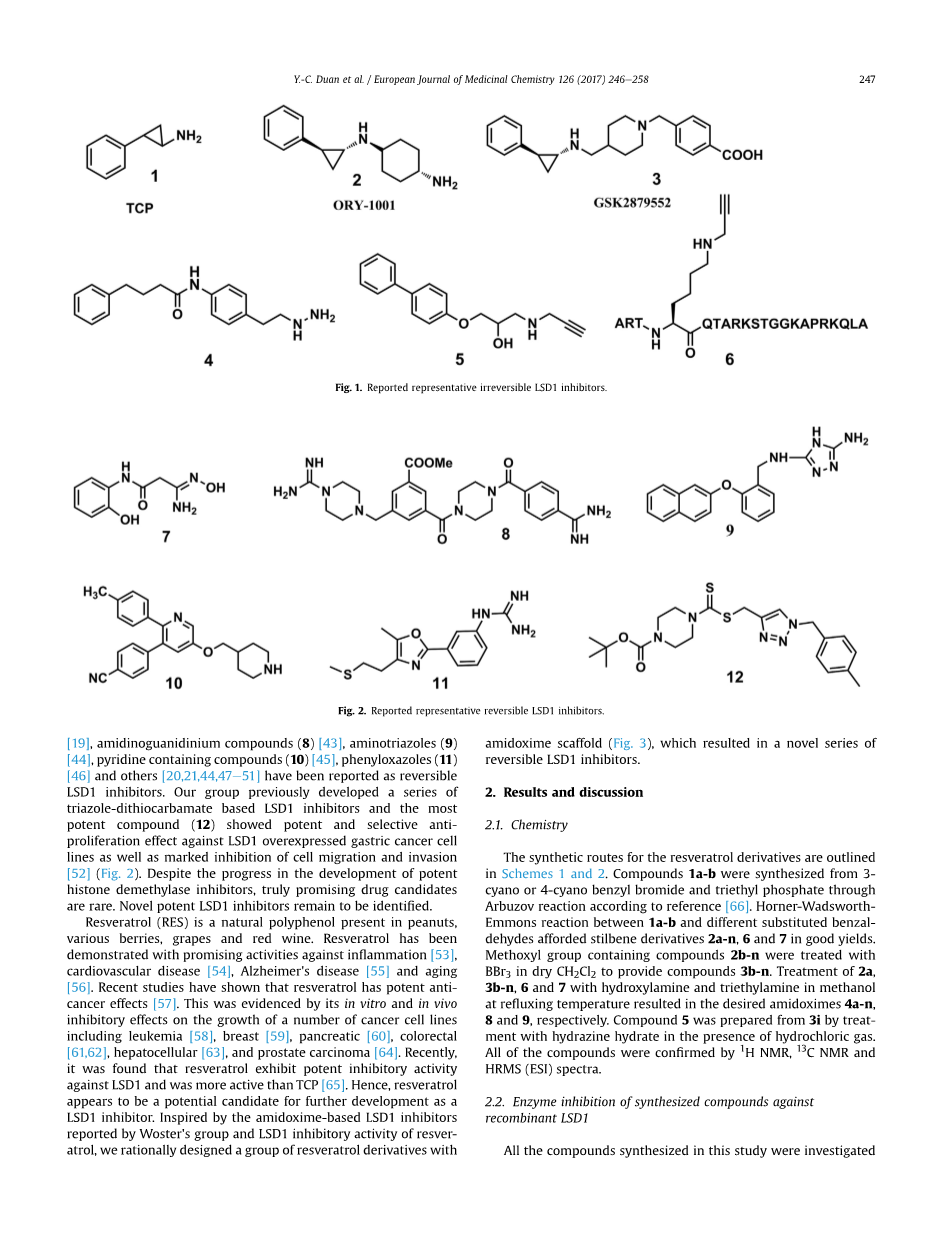
自2004年发现LSD1以来,已经确定了许多不同的LSD1抑制剂分支[30-32]。研究槜广泛的和最有效的LSD1抑制剂是不可逆的,而且是基于著名的抗MAO试剂:强内心百乐明(用以抑制单胺氧化酶的抗抑郁药)(TCP,1)[33-38](图1)来设计的。直到现在,两种基于三氯苯酚的LSD1抑制剂ORY-1001(2)和GSK2879552(3),正在进行癌症治疗的临床试验[39,40]。其它LSD1不可逆抑制剂正着重于苯乙嗪-(4)[41]和炔丙基胺基化合物(5,6)[17,42]的发展。最近,强有力的、类药物的和可逆的小分子LSD1抑制剂的开发引发兴趣热潮。各种不同的抑制剂,包括偕胺肟基化合物(7)[19],脒基胍化合物(8)[43],氨基三唑(9)[44],含吡啶化合物(10)[45]。苯基噁唑(11)[46]和物质[20,21,44,47-51]都已被报道为可逆的LSD1抑制剂。我们团队以前开发的一系列基于三唑-二硫代氨基甲酸酯的LSD1抑制剂和最有效化合物(12)都对LSD1高表达胃癌细胞株具有明显的抗增殖作用,对细胞迁移和侵袭有明显的抑制作用[52](图2)。尽管在开发有效组蛋白去甲基化酶抑制剂方面取得了进展,但真正有希望的候选药物却很少。新的有效LSD1抑制剂仍有待鉴定。
图1:报道的代表性不可逆LSD1抑制剂
图2:报道的代表性可逆LSD1抑制剂
白藜芦醇(RES)是花生,各种浆果,葡萄和红酒中存在的一种天然多酚。白藜芦醇已被证明对于对抗炎症[53],心血管疾病[54]、阿尔茨海默病[55]和衰老[56]具有很好活性。最近的研究表明白藜芦醇有很强的抗癌作用[57]。通过其对包括白血病[58]、乳腺[59]、胰腺[60]、结直肠[61、62]、肝细胞[63]和前列腺癌[64]在内的多种癌细胞系的生长的体外和体内抑制作用证明了这一点。最近,发现白藜芦醇对LSD1具有强抑制活性,且比TCP[65]更具活性。因此,白藜芦醇作为LSD1抑制剂有望进一步发展。受Woster团队报道的偕胺肟类LSD1抑制剂和白藜芦醇LSD1抑制活性的启发,我们合理地设计了一组带有偕胺肟支架的白藜芦醇衍生物(图3),得到了一系列新的可逆LSD1抑制剂。
图3:本文设计合成了新型LSD1抑制剂
2.结果和讨论
2.1化学
白藜芦醇衍生物的合成路线概述在方案1和2中。化合物1a-b由3-氰基或4-氰基苄基溴和磷酸三乙酯通过熊果苷反应合成,参考文献[66]。1a-b与不同取代苯甲醛的霍纳尔-沃兹沃思-埃蒙斯反应反应得到二苯乙烯衍生物2a-n、6和7,产率较高。甲氧基含化合物2b-n混合BBr3在干燥的CH2Cl2中合成化合物3b-n。在回流温度下用羟胺和三乙胺在甲醇中处理2a、3b-n、6和7分别得到所需的脒肟4a-n、8和9。化合物5由3i在盐酸存在下用水合肼处理制备。所有化合物均经1HNMR、13CNMR和电喷雾质谱(ESI)表征。
方案1:化合物2-5的合成。试剂和条件:(a)二甲基甲酰胺,叔丁醇,0℃-rt,0.5-2h:(b)BBr3,二氯甲烷,-35℃-rt,过夜;(c)NH2OH﹒HCl,Et3N、CH3OH、回流,3-5h;(d)(i)3i,乙醇,盐酸(气体),0℃-rt,2h;(ii)NH2NH2﹒H2O,乙醇,室温,3h.
方案2:化合物8-9的合成。试剂和条件:(a)1a、叔丁醇,叔丁醇钾盐,0℃-rt,1h;(b)1b、二甲基甲酰胺,叔丁醇,0℃-rt,2h;(c)NH2OH﹒HCl,Et3N,CH3OH,回流,5h.
2.2合成化合物对重组酶LSD1酶的抑制作用
对合成的化合物进行体外抑菌活性研究。三氯丁二烯和苯乙烯分别为作为阳性对照。如表1所示,除了化合物4h和4n之外,所有化合物都表现出中等至强的抑制LSD1活性。其中,在苯环上具有羟基取代基的化合物4b-4g和4i-4m具有较强的抑制活性,其IC50值在121nM至2.59mM之间,比TCP和RES都强。其中4e(IC50121.23nM)和4m(IC50123.86nM)具有最强的抗LSD1活性,是RES的80倍。与化合物4j或4i相比,无羟基取代基的化合物4a在10mm处对LSD1的抑制率仅为48.9%,但化合物4h和4n活性缺失的原因尚不清楚。用吡啶环(4bvs8)或吲哚环(4ivs9)取代苯基支架导致活性显著降低,这表明苯环在保持其活性方面具有一定意义。通过比较4i和5,将苯并咪唑酰胺转化为苯并咪唑酰肼显著降低了抑制活性。间氨基肟取代是优选的:间氨基肟取代的衍生物(4b-4g)比相应的对氨基肟取代的衍生物对抗LDS1更有效。
表一:化合物4a-n、5、8和9的体外LSD1抑制活性
接下表:
a.数据表示为10mM或IC50值时的抑制百分比(平均SD)。所有实验独立进行至少三次。
b.未测试。
c.无抑制作用。
2.3可逆性研究
为了测试4e(图4A)和4m(图4B)的LSD1的可逆性,我们使用稀释测定法。并发现,将LSD1/化合物混合物稀释80倍可使LSD1活性恢复。然而,在共价结合抑制剂GSK2879552[67]的存在下,稀释后不能恢复LSD1活性。这些结果表明4e和4m与GSK2879552相比具有可逆性,这意味着4e和4m能够以非共价方式与LSD1重组体结合。
图4:稀释法测定4e(A)和4m(B)对LSD1活性的可逆性,以GSK2879552为对照。数据是平均标准差。误差考虑为**plt;0.01,差异有统计学意义。所有实验至少进行三次。
2.4 4e和4m对MGC-803细胞的抑制作用
为了进一步确认4e和4m的细胞活性,将不同浓度的4e和4m应用于过度表达的LSD1[68]的胃癌细胞系MGC–803中。如图5所示,将MGC-803细胞接种在96孔板中,并用这些抑制剂处理5天。然后用H3K4me2抗体对细胞进行免疫荧光,以指示细胞中LSD1的活性。同时,DAPI也用于细胞计数(图5)。用高含量分析的12目免疫荧光分析每个孔,评价H3K4me2的强度,H3K4me2的强度与细胞数的比值指示LSD1的活性。从图6可以看出,当用4e(图6E)和4m(图6F)处理细胞三天时观察到4e(图6A)和4m(
剩余内容已隐藏,支付完成后下载完整资料

英语原文共 13 页,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[281109],资料为PDF文档或Word文档,PDF文档可免费转换为Word
以上是毕业论文外文翻译,课题毕业论文、任务书、文献综述、开题报告、程序设计、图纸设计等资料可联系客服协助查找。
您可能感兴趣的文章
- 合成与光催化从新型二氧化铈和掺银二氧铈中光解水制氢静电纺丝法制备纤维外文翻译资料
- 利用2-苯基-1h-咪唑基丹参酮IIA衍生物选择性稳定多个启动子g-四重体DNA及其在转移性乳腺癌中的潜在抑制作用外文翻译资料
- 直接芳基化反应和非均相催化的结合外文翻译资料
- “顺势疗法”钯纳米颗粒催化交叉碳–碳偶联反应外文翻译资料
- 磁性纳米粒子负载离子改性TBD:一种应用于有机转换的高效、可回收催化剂外文翻译资料
- 扩展的紫罗碱综合环芬CdS量子点中的超快的两电子转移外文翻译资料
- 实现一个重要明显增加EFfi效率在相应的纯蓝色荧光OLED:准等价的杂化态外文翻译资料
- 基于咪唑-π-三苯胺衍生物的高效深蓝色有机发光装置外文翻译资料
- 从3-氨基-1-丙醇或3-卤丙胺氢卤化物出发,通过n -三硝基或n -二甲氧基三苄基lazetiine高效合成氮叠丁外文翻译资料
- 气相色谱引样方法外文翻译资料

