
英语原文共 5 页,剩余内容已隐藏,支付完成后下载完整资料
添加硅对由含聚碳硅烷涂层B4C粉末制备的B4C-SiC复合材料的微观结构和性能的影响
摘 要
采用热压热解法由聚碳硅烷涂覆的B4C粉末和Si添加剂在1950℃下在30MPa下1小时的混合成功制备了致密的细晶B4C-SiC复合材料。Si的加入不仅改善了由于液相Si的形成而致密化过程,而且还大大提高了B4C陶瓷的力学性能。 然而,添加的Si对断裂韧性没有显着影响。 机械性能的改善是归因于孔隙率的降低和来自聚碳硅烷和B4C粉末的游离碳的消除。 Si的引入进一步改善了微观结构的均匀性,改善了原位SiC晶粒的层状结构。用11.4wt%Si制备的样品其弯曲强度,维氏硬度和断裂韧性达到389MPa,33.2GPa,和5.64 MPa m1/2。
关键词:B4C 碳化硅 聚碳 热压 机械性能
1. 简介
作为一种很有前途的材料,碳化硼(B4C)已被用于研磨材料,吸收性核材料,喷射喷嘴,和高温热电转换材料。其优异的性能,如高硬度(430 GPa),高熔点(2450℃),中子容量高,横截面积大,化学稳定性好[1,2]。特别是它高硬度和极低密度(2.52克/立方厘米)对装甲应用和摩擦学应用非常有吸引力。在这两个应用中,备受关注和努力致力于同时增强密度-陶瓷的阳离子,硬度和韧性[3]。但是,它广泛的应用受到其不良骨折的限制由于B的强共价键,韧性和可烧结性和低自扩散系数[4]。为了减轻或克服上述缺点,已经开发出新的流程和方法改善烧结性能,通过添加可以改善B4C陶瓷烧结添加剂,如金属[5,6]和金属氧化物[7-10],原因是能在烧结过程中形成液相。这些添加剂可能有益于致密化,但它们的副作用不容忽视,特别是劣化了高硬度的独特性能。在保持高硬度的同时提高B4C韧性的解决方案仍然是一个挑战。以前的调查表明了用聚碳硅烷(PCS)作为SiC的来源的制备方法来改善烧结行为和微观结构是可以接受的[11-13]。在我们以前的工作中,通过热压热解涂有PCS [14]B4C粉末混合物制备B4C-SiC复合材料成功了。但是,热分解聚碳硅烷通常不仅产生SiC,还产生游离碳。在
无压烧结,来自PCS的碳杂质可以对B4C的致密化有积极作用能减少碳化硼粉末的氧化层[15],但是因此,烧结机理不同在热压过程中不会产生影响。同时,应该指出游离碳杂质会降低强度,硬度,和严重影响复合材料的抗氧化性 [16]。在该论文中,B4C-SiC复合材料是用聚碳硅烷涂覆的B4C粉末和Si添加剂的热压热解混合物制造的。Si添加剂对用聚碳硅烷涂覆的B4C粉末制备的B4C-SiC烧结韧性,微观结构和力学性能的影响复合材料已被研究。
2. 实验过程
2.1 原料和方法
研究中使用的起始粉末包括B4C(中国牡丹江市牡丹江江静安碳化硼有限公司),PCS(平均分子量1300),国立大学中国长沙国防科技有限公司的高纯硅。根据供应商的数据,B4C的平均粒径粉末为3.5mu;m。 PCS的热解固体产率约为60%。热解残留物由85.4wt%的SiC和14.6wt%的游离碳。计算PCS的量以获得含有15wt%(SiCthorn;C)的复合粉末。为了转换将碳释放到SiC中,添加不同含量的Si粉末加入复合粉末。
首先,将PCS溶解在无水环己烷中。然后是将B4C粉末和Si粉末分散在PCS溶液中进行8小时激烈的搅拌。通过旋转蒸发除去溶剂,在70℃,然后将混合物干燥24小时。筛分后将干燥的混合物通过200目筛,将聚碳硅烷涂覆的粉末在850℃下在管式炉中热解2小时并用Ar气氛保护。球磨2小时后,然后将得到的粉末通过200目筛子筛分,填充到石墨模具中进行热压。混合粉末首先在真空下从室温加热到1500℃压力,然后在1500℃保持30分钟。将压块在Ar气氛中连续加热至1950℃并在烧结温度下和30兆帕下保持1小时。
2.2 过程描述
在去离子水中测量烧结样品的密度,根据阿基米德方法三次测量并进行干燥,悬浮和饱和权重。 将样品在去离子水中煮沸1小时,然后在测量悬浮和冷却之前冷却至室温饱和重量。 样品的结晶相是相同的。通过X射线衍射(XRD; Rigaku Ultima RINT2100,Japan)进行测试。
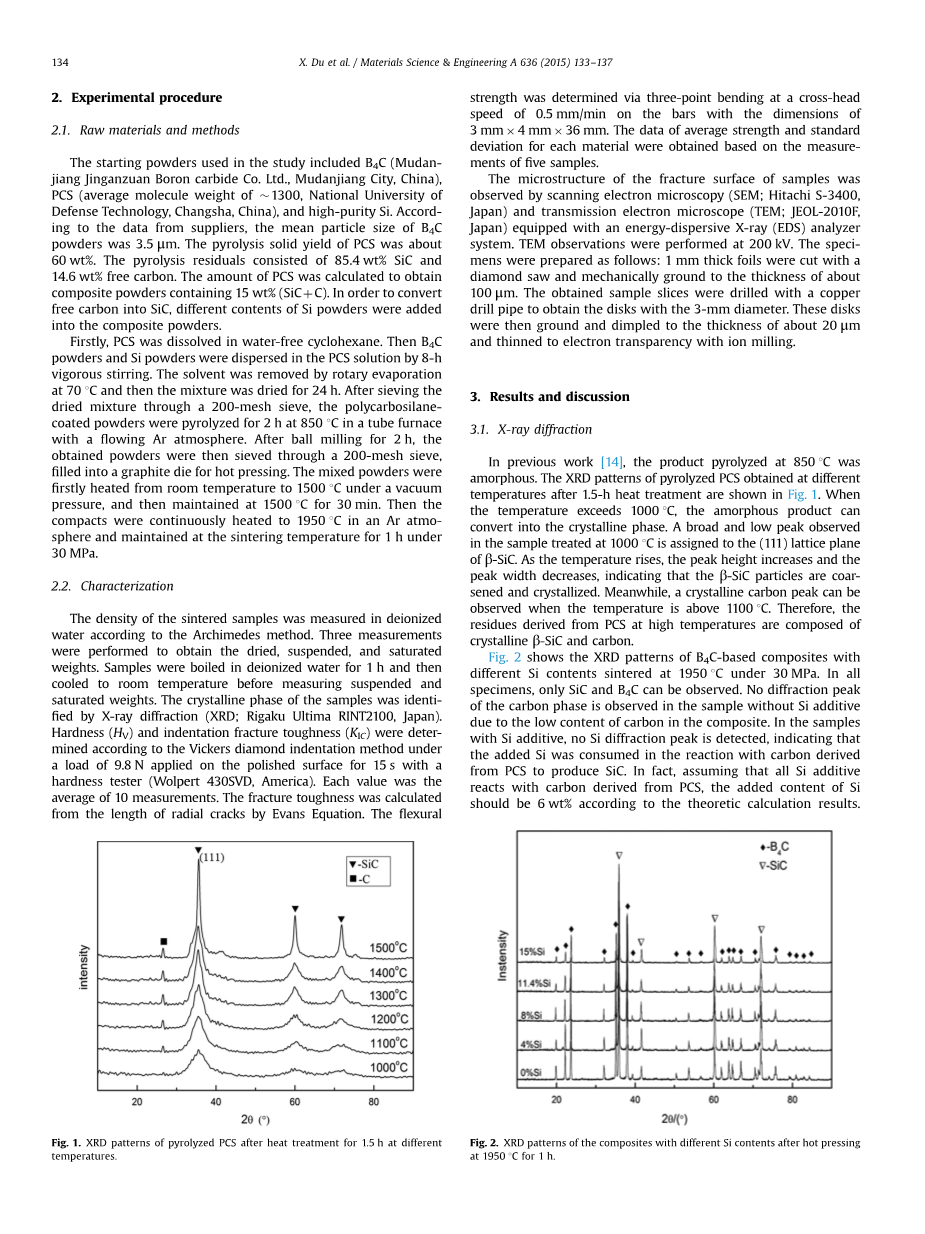
图1 PCS热处理1.5h后不同热处理温度下的XRD图谱
根据Vickers金刚石压痕法确定硬度(HV)和压痕断裂韧性(KIC)在抛光表面上施加9.8N的负载15秒硬度计(Wolpert 430SVD,America)。 每个值都平均10次测量。由埃文斯方程的径向裂缝长度计算断裂韧性。通过十字头处的三点弯曲确定弯曲强度,棒尺寸为0.5 mm/min,尺寸为3毫米4毫米36毫米。基于五个样品的测量获得每种材料的偏差平均强度和标准的数据。样品断裂面的微观结构则通过扫描电子显微镜观察(SEM; Hitachi S-3400,日本)和透射电子显微镜(TEM; JEOL-2010F,日本)配备能量色散X射线(EDS)分析仪系统。TEM观察在200kV下进行。如下制备特定样品:用1mm切割1mm厚的箔金刚石锯并机械研磨至约厚度100微米 获得的样品切片用铜钻孔钻杆以获得直径为3毫米的圆盘。然后将其研磨并凹陷至约20mu;m的厚度并用离子铣削稀释到电子透明度。
3. 结果与讨论
3.1 X射线衍射
在以前的工作[14]中,产物在850℃下1.5小时热处理后的热解得到的热解PCS的XRD图谱如图1所示。温度超过1000℃,无定形产品即可转化为结晶相。 观察到广泛的低峰,在1000℃处理的样品中,将(111)晶格平面分配给(111)
beta;-SiC。 随着温度的升高,峰值高度增加峰宽减小,表明beta;-SiC颗粒是煅烧和结晶的。同时,结晶碳峰可以当温度高于1100℃时观察到。 因此,在高温下由PCS衍生的残留物由结晶beta;-SiC和碳组成。
图2 1950摄氏度持续1小时不同硅含量复合材料热压后的XRD图谱
图2显示了基于B4C的复合材料的XRD图谱。不同的Si含量在1950℃,30MPa下烧结。在所有样品中,只能观察到SiC和B4C。没有衍射峰在没有Si添加剂的样品中是由于复合材料中碳含量低。 在样本中用Si添加剂,没有检测到Si衍射峰,表明添加的Si在与碳衍生的反应中被消耗从PCS生产SiC。 实际上,假设所有的Si添加剂与来自PCS的碳反应,Si的添加量根据理论计算结果,应为6wt%。
但是,在目前的工作中,即使Si的添加量达到了11.5wt%,未检测到Si峰,表明除游离外来自PCS的碳,原料B4C粉末也含有少量游离碳的量。 另外,SiC峰值强度增强和B4C峰强度的降低表明了增加复合材料中的SiC含量并确定其原因是缺少Si。
3.2 微观结构分析
为了进一步研究相组成,分析进行EDS。图3显示了样品的TEM图像。用15wt%Si和相应的EDS光谱。在三角形晶粒连接处观察到轻微残留的Si相。值得注意的是,B-C-Si晶界附近的三元碳化物相也可以检测到,表明Si在B4C中溶解。泰尔和佩佐[17]报道了在B4C-B-Si系统中Si在B4C中的溶解,低压烧结Si在B4C中的固溶度约2.5%。 Frage [18,19]研究了热力学过程,在三元B-C-Si系统和微观结构演变在熔融硅和碳化硅的渗透过程中发现溶解-沉淀机制可能是负责形成B-C-Si三元碳化物层。Si在B4C基质中的溶解促进了质量传递,在B4C陶瓷的固相烧结中对研究的Si掺杂复合材料的致密化也有部分贡献。
具有不同Si含量的复合材料热压B4C断裂面的SEM图像如图4所示。可见在没有Si添加剂的样品中观察到孔隙,表明密度相对较低。 随着Si粉末的添加量
复合材料中的孔隙明显减少。当添加的Si粉末的含量超过8wt%的时候,样品中的孔隙几乎观察不到,毛孔的形状也变成了球形,表明添加Si确实促进了B4C基陶瓷的致密化。 同时,纳入Si有利于微观结构的均匀性。 B4C-SiC复 合材料的微观结构受体积和原位SiC颗粒的比例的影响。在图4中,深灰色区域是B4C相,小白色区域是SiC相。白色细晶粒SiC相均匀分布在深灰色B4C基质中并抑制B4C颗粒的生长。 样品中含Si添加剂B4C颗粒的平均尺寸小于5mu;m,表明晶粒尺寸几乎没有成长。 虽然Si的引入有益于SiC晶粒的生长,原位SiC的平均晶粒尺寸仍然不超过2mu;m,而SiC晶粒尺寸的分布是均匀而狭窄的。 聚集现象观测不到。
图3 spot模式下15 wt%Si样品的TEM图像和相应的EDS谱
图4 不同硅含量试样断口的扫描电镜照片:(a)0 wt%Si;(b)4 wt%Si;(c)8 wt%Si;(d)11.4 wt%Si;(e)15 wt%S
图5 不同加硅量复合材料相对密度和硬度的变化
在不含Si添加剂的样品中,B4C的平均晶粒尺寸晶粒大于含有Si添加剂的样品。在我们的以前的工作[14],大多数来自PCS的SiC颗粒都是位于B4C晶界处,但是位于尺寸范围小于300nm的一些SiC纳米颗粒或准纳米颗粒在B4C晶粒内,表明了内部/类型间微观结构。通常,较小的SiC颗粒作为钉扎位点的效果较差,因为它们很容易被快速移动的晶界所吞没,倾向于在粒内位置结束。但是,晶界更大的SiC颗粒倾向于更有效地钉扎,随着晶界移动,被拖动。显然,B4C基体晶粒的细化应归因于SiC晶粒在B4C晶界之间,可以固定迁移的晶粒边界并抑制B4C颗粒的生长。因为高SiC颗粒的体积分数可以增加SiC-B4C的总面积晶界,晶界的能量需求扩散和晶界迁移率发生变化。 Piciachio [20]和Parchoviansky [21]报道了在A2O3-SiC复合材料中的应用,SiC颗粒对Al2O3晶粒生长的抑制作用随着SiC体积分数的增加而放大。因此,这种精细且均匀的微观结构表明添加Si确实改善了复合材料的微观结构。
3.3 机械性能
图5显示了具有不同Si含量的复合材料B4C的相对密度曲线和硬度曲线。由阿基米德方法得到的密度结果与B4C复合材料断裂面的显微照片结果非常一致。随着Si含量的增加,相对密度增加到11.4%(重量)。进一步添加至15%,略微降低。没有Si添加剂的B4C复合材料的密度约为95%。后加入Si粉末和热压烧结,相对的复合材料的密度进一步提高。复合材料11.4wt%的Si几乎完全致密,相对密度为99.2%。众所周知,硅粉熔化在1410℃以上。很明显,复合材料中的致密化增强是由于形成液态Si相。当添加的Si高于11.4wt%时,相对密度的轻微下降可能是由于高温下过量液相Si的剧烈挥发。
类似于相对密度,研究B4C复合材料硬度具有相同的趋势。硬度在没有Si添加剂的样品仅约24 GPa,但含11.4%(重量)Si的样品可达到33.2 GPa,即高于38%,对于不含Si添加剂的样品。显然,硬度的增加是由残余孔隙率的降低引起的,在样本中。一般来说,复合材料的硬度主要是取决于孔隙率,固有硬度和体积含量。对于具有相同成分的样品,残余孔隙率越低意味着硬度越高。在相对密度相同的样品中,组分的硬度随着第二相的体积含量的增加而增加。众所周知碳作为软相会使其机械性能变差。在论文中,除了残留的毛孔,晶界中的残余碳也可能是造成的没有Si添加剂的样品硬度相对较低,因为陶瓷中存在富碳相在压痕过程中导致严重的塑性变形并导致硬度降低[16]。因此,增加了硬度是由软碳的去除和SiC的形成引起的。当Si添加剂的含量进一步增加到15wt%,样品的硬度略有下降。因此,引入Si添加剂不仅由于液相的形成促进了致密化,在烧结过程中,通过去除富碳相也提高了B4C陶瓷的硬度。
Si含量对弯曲强度和断裂的影响韧性如图6所示。抗弯强度首先降低,当Si含量高达4%(重量)时,则略有增加。将Si的量增加至11.4wt%,开始减少。当它进一步增加到15%(重量)时,再次略微增加。 相比之下没有Si添加剂的样品,复合材料的抗弯强度随着Si的高含量显着提高。 样本用11.4wt%Si表现出389MPa的最高挠曲强度,而没有Si添加剂的样品仅为265MPa。 根据
格里菲斯理论,原则上两个主要因素提高陶瓷材料的抗弯强度。一个是减少了先前存在的缺陷或微裂纹的大小。另一个是增加裂缝的能量障碍传播。显然,含Si添加剂的样品弯曲强度的提高归因于相对密度的增加和孔隙缺陷的减少与高密度。值得注意的是,具有4wt%Si的样品的强度略低于不含Si添加剂的样品。它可能是由不均匀的微观结构和缺陷导致的开裂毛孔造成的,这容易导致应力集中毛孔尖端。而且,Si的添加可以细化原位SiC晶粒的层结构。
图6 研究对象的弯曲强度和断裂韧性的变化
图7 SiC颗粒的TEM显微照片:(a)未添加Si的样品;(b) 含有11.4 wt%Si的样品;(c)局部区域的HTEM图像,对应于(b)中的框区域。
图7显示了SiC颗粒的典型TEM图像。可以看出,SiC颗粒具有层结构。对于不含Si添加剂的样品,层结构排列成非常整洁有序的方式,层之间的距离是差不多一样。但是,在具有11.4wt%Si的样品中,该层可以观察到具有不等层间距的结构。一层厚厚的可以被分成许多薄层,这些薄层可以相互作用,导致层结构局部无序。可以将SiC晶粒中的薄层结构归因于溶解-沉淀过程。根据Pampuch[22]和Ness和Page [23]等人的研究结果,在最初在相互作用阶段,碳溶解在硅熔体中然后扩散到SiC颗粒的表面和非均相沉淀,新的SiC发生并始终以手指状颗粒开始。该精细的异质层结构可能是一个有效的障碍阻止裂纹扩展,因为这些局部无序区域在层中,当裂纹扩展
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[240954],资料为PDF文档或Word文档,PDF文档可免费转换为Word
您可能感兴趣的文章
- y掺杂Li8ZrO6:一种高容量锂离子电池正极材料外文翻译资料
- 水泥基灌浆材料对半柔性沥青路面性能的影响外文翻译资料
- 溶胶-凝胶法制备不同Eu3 掺杂含量的Al2O3的结晶和发光性质外文翻译资料
- 溶胶-凝胶法制备的掺杂有Eu3 的氧化铝的结晶和发光性能外文翻译资料
- 具有高圆偏振光致发光的手性二维钙钛矿外文翻译资料
- N-杂环卡宾催化的对映选择性环化反应外文翻译资料
- 有机催化不对称N-磺酰基酰胺C_N钡活化以获 取轴向手性联芳基氨基酸外文翻译资料
- 用于钠离子存储的空心Mxene球体和三维多孔MXene结构.外文翻译资料
- 深共熔溶剂中铜基Sn-Co-Ni和Sn-Co-Zn合金镀层的电沉积及其表征外文翻译资料
- 工作在类合金模型的两个非富勒烯受体,使三元有机 太阳能电池的效率超过17%外文翻译资料

