
英语原文共 12 页,剩余内容已隐藏,支付完成后下载完整资料
一种全固态可充电氯离子电池
Chao Chen, Tingting Yu, Meng Yang, Xiangyu Zhao,* and Xiaodong Shen
- 介绍
氯离子电池是继锂离子电池之后发展起来的一种新型电池材料,具有丰富的材料资源和较高的能量密度。然而,由于电极材料的溶解和在液体电解质中的副反应,限制了它的应用。本文报道了一种允许氯离子转移的固体聚合物电解质,它由聚环氧乙烷为聚合物基体,三丁基甲基氯化铵为氯盐,丁二腈为固体增塑剂组成。所制备的聚合物电解质在298-343k的温度范围内具有10-5-10-4s cm-1的导电性,当与氧化铁/锂电极系统组装时,实现了FeOCl/FeO在阴极和Li/LiCl在阳极的可逆电化学氧化还原反应,下面演示第一个全固态可充电氯离子电池
随着化石能源的日益短缺和环境的污染,发展可持续的清洁能源及其收获、转化、储存和利用成为人们关注的焦点可充电电池被认为是一种典型的储能技术,在便携式电子设备、移动仪器和固定的电网规模站中有着不同的应用除铅酸电池、[5]镍氢电池、[6]锂离子电池等商用电池外,[7]各种基于阳离子(Na 、K 、Mg2 、Ca2 、Zn2 、Al3 )的替代电池化学,阴离子(F-,Cl-,O2-),[14–18]或双离子(Li /TFSI-,AlCl4-/Al3 、Ca2 /PF6-、Li /Mg2 ;TFSI双(三氟甲基磺酰)酰亚胺的转移研究越来越多。关于阴离子电池系统,氯离子电池(CIB)因其无毒、富含氯化物的电极材料和电解液材料在世界范围内广泛应用而备受关注。此外,CIB还显示了多种潜在的电化学偶,其理论体积能量密度高达2500wh l-1,高于传统锂离子电池
C. Chen, Dr. T. Yu, Dr. M. Yang, Prof. X. Y. Zhao, Prof. X. D. Shen
南京理工大学材料科学与工程学院江苏先进无机功能复合材料合作创新中心
中国南京211816 电子邮件:xiangyu.zhao@njtech.edu.cn
X. D. Shen教授
南京理工大学材料重点化学工程国家重点实验室
本文作者的ORCID标识号可在https://doi.org/10.1002/advs.201802130下找到。
版权所有2019作者。由威利VCH Verlag GmbHamp;Co.KGaA,Weinheim出版。这是《知识共享署名许可证》条款下的开放获取文章,允许在任何媒体中使用、分发和复制,前提是正确引用了原始作品。DOI: 10.1002/advs.201802130
CIB的概念首先是以路易斯酸金属氯化物为阴极,碱金属或碱金属为阳极,二元离子液体为电解液,将氯离子液体溶解在具有类似有机阳离子和另一阴离子如BF4-或TFSI-的其它离子液体溶剂中而实现的。然而,金属氯化物阴极在这种液体电解质中的溶解和穿梭导致CIB的容量严重衰减。另一种方法是使用金属氯氧化合物材料,如BiOCl和FeOCl或掺杂氯离子的高稳定性导电聚合物材料作为阴极,以避免这种溶解问题。然而,与金属氯化物阴极相比,这些新阴极材料显示出能量密度降低。在正极材料方面,采用与液体电解质相容性好的金属锂作为CIBs的负极材料,对其进行了研究。注意,CIBs的一个显著优点是可以使用大量的材料,如Na、Mg和Ca作为阳极。[30]采用金属氯氧化合物/Mg或金属氯氧化合物/Mg-MgCl2电极体系,证明了镁阳极在CIBs中的可行性,虽然采用了镁复合阳极材料,但其电化学性能不如锂金属阳极体系。[30,31] 这种不良性能可能是由于液态电解液中阳极侧的MgCl2放电产物溶解,以及液态电解液中的金属镁和溶剂的大TFSI-阴离子之间的副反应[31,32]可在镁金属表面分解,在循环过程中堵塞镁。此外,二元离子液体电解质中的杂质也可能使镁阳极变质。
解决电极溶解和电极材料与溶剂/液体电解质之间的副反应问题的一种可能方法是使用固体电解质,这在锂离子电池和金属硫电池中被证明是非常有效的。[33-35] 然而,关于固体氯离子导体的研究还很少。具有较高电化学稳定性的无机氯离子导体,如BaCl2、SrCl2、LaCl3和LaOCl多晶材料,需要500 K以上的高温才能获得约10-6 S cm-1的离子导电性。[36,37] 一些其他二元金属氯化物,如PbCl2基材料,在室温下具有高于10-5s cm-1的离子导电率,[38,39],但其相关的化学稳定性较低。作为钙钛矿型氯化物之一的三元立方CsSnCl3材料在室温下获得了10-4s cm-1的较高离子电导率。[39,40]然而,由于CsSnCl3的半导体特性,不可能将其用作电解质。[40] 以准固态高分子材料为基础的有机氯离子导体也有报道。Hardy和Shriver开发了一种以液态聚乙二醇(PEG)为氯离子导电聚合物电解质增塑的聚(二烯丙基二甲基氯化铵)(PDDAC),室温下电导率为10-5s cm-1,[41],通过添加季铵盐可以将其增加到10-4s cm-1。[42] 类似的概念也被用于制备由聚氯乙烯、明胶或聚二氟乙烯六氟聚合物、季铵盐和甘油、聚乙二醇或邻苯二甲酸二正戊酯的液体增塑剂组成的准固态聚合物电解质。[43] 这种电解液被用来制造电池,但是基于氯离子转移的电化学反应证据很弱。组装后的高含水量电池会引起严重的化学氯离子腐蚀。此外,未提供组装电池的可充电性,仅显示第一次放电行为。
- 结果和讨论
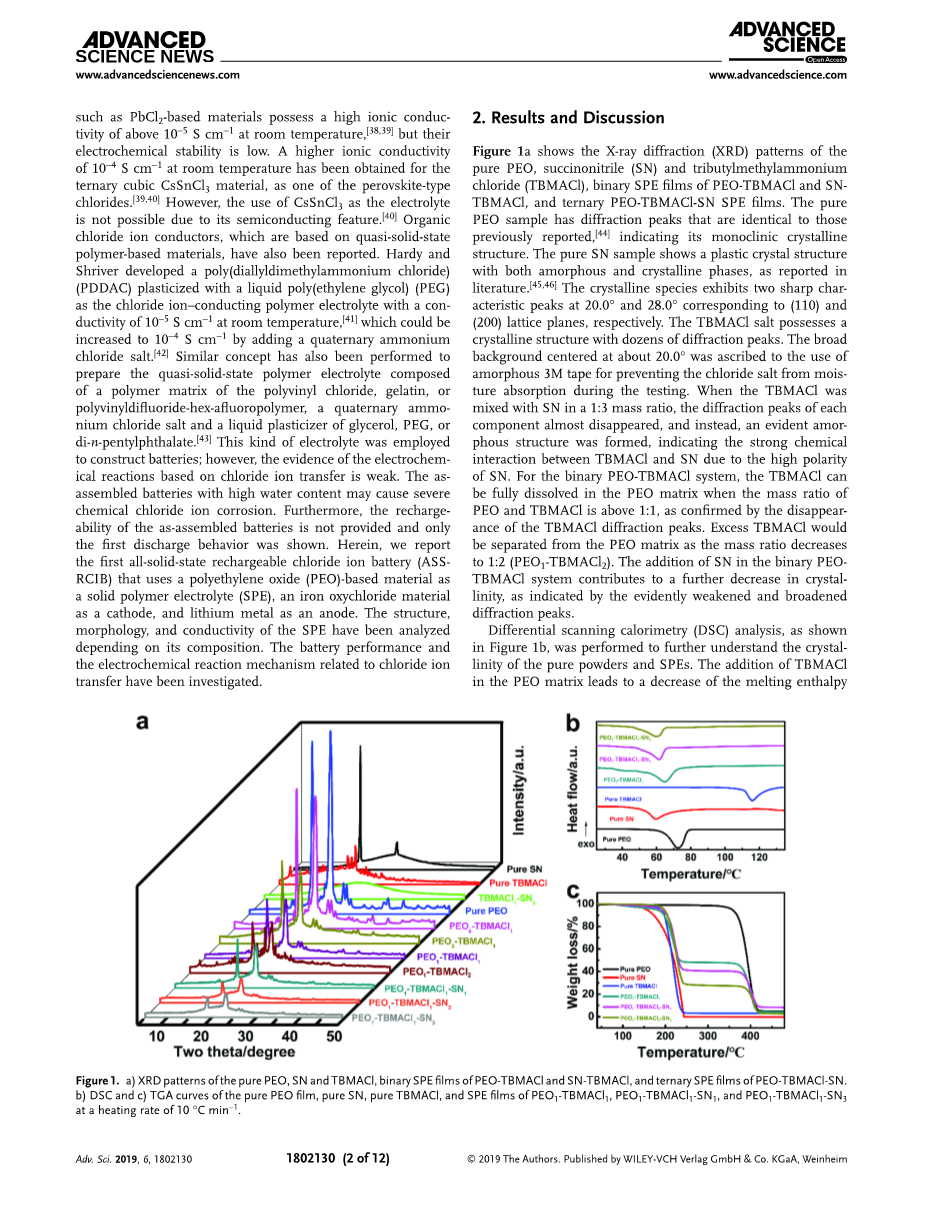
图1a显示了纯PEO、丁二腈(SN)和三丁基甲基氯化铵(TBMACl)的X射线衍射(XRD)图、PEO-TBMACl和SNTBMACl的二元SPE膜以及PEO-TBMACl-SN的三元SPE膜。纯PEO样品的衍射峰与先前报道的相同[44],表明其单斜晶体结构。如文献所述,纯SN样品具有非晶和晶相的塑性晶体结构。[45,46] 晶体在20.0°和28.0°分别出现两个与(110)和(200)晶格面对应的尖锐特征峰。TBMACl盐具有晶体结构,有几十个衍射峰。以约20.0°为中心的宽背景归因于在试验期间使用非晶态3M胶带防止氯盐吸湿。当TBMACl与SN以1:3的质量比混合时,各组分的衍射峰几乎消失,形成了明显的非晶态结构,表明TBMACl与SN之间由于SN的高极性而发生了强烈的化学作用。对于二元PEO-TBMACl体系,当PEO与TBMACl的质量比大于1:1时,TBMACl完全溶解在PEO基体中,TBMACl衍射峰的消失证实了这一点。当质量比降低到1:2(PEO1-TBMACl2)时,多余的TBMACl将从PEO基质中分离出来。二元peotmbacl体系中SN的加入使结晶度进一步降低,衍射峰明显减弱和展宽。
图1。A)纯PEO,SN和TBMACl的XRD图谱,PEO-TBMACl和SN-TBMACl的二元SPE薄膜,以及PEO-TBMACl-SN的三元SPE薄膜。
B)纯PEO薄膜、纯SN薄膜、纯TBMACl薄膜和PEO1-TBMACl1-sn1、PEO1-TBMACl1-sn1、PEO1-TBMACl1-sn3薄膜的DSC和TGA曲线。
进行差示扫描量热(DSC)分析,如图1b所示,以进一步了解纯粉末和spe的结晶度。TBMACl在PEO基体中的加入导致熔融焓从174.0(PEO)降低到134.4j g-1(PEO1-TBMACl1),表明结晶度降低,随着SN的引入结晶度不断降低。PEO1-TBMACl1-SN1和PEO1-TBMACl1-SN3的熔融焓分别为103.2和84.2j g-1。熔点(即吸热峰的中点)也有相似的变化趋势,从72.6(PEO)降低到59.3℃(PEO1-TBMACl1-SN3)。图1c显示了相应的热重分析(TGA)。PEO基体在340℃高温下具有良好的热稳定性,加入50wt%TBMACl(PEO1-TBMACl1)后热稳定性提高到370℃。在纯样品和二元PEO1-TBMACl1固相萃取中,TBMACl显示出大约180℃的相似热稳定性。纯锡的热稳定性最低,在120℃左右开始减重,这与之前的报告相似。[45] 注意,三元PEO-TBMACl-SN-SPE在180℃左右开始急剧失重,这说明SN的热稳定性是由复合SPE中的化学相互作用提高的,这将在以后进一步分析。
支持信息中的图2和图S1显示了光学照片、扫描电子显微镜图像(SEM),PEO和PEO-TBMACl-SN-SPE薄膜的元素对应图。PEO和PEO1-TBMACl1-SN3样品具有相似的光学形貌,结构完整,柔韧性好。PEO1-TBMACl1-SN3 SPE薄膜(图S1b,支持信息)的表面更加光滑,其厚度约为80mu;m(图2c),C、O、N和Cl元素分布均匀,表明SN和TBMACl在PEO基体中分散良好。随着SN含量的增加(PEO1-TBMACl1-SN4),制备的SPE膜失去了结构完整性,在膜中形成了许多孔洞(图S1c,h,i,支持信息)。这是由于真空干燥过程中三元SPE膜中过量SN的挥发造成的。进行傅里叶变换红外光谱(FTIR)以进一步阐明SPE膜中PEO、TBMACl和锡组分的相互作用,如图3所示。支持信息中的表S1-S3列出了这些纯组件的特性分配。TBMACl1-SN3混合物由于出现属于TBMACl和SN的带而呈现出双相性质。但观察到明显的峰移。与SN[47]的CH2伸缩振动有关的2990和2952cm-1处的谱带在加入TBMACl后向较低波数(红移)移动超过15cm-1。对应于CH2行波振动和摇摆振动的谱带显示出类似的位移。同时,发生了-Cequiv;N的电子损失,[48]导致氰化物峰从2257红移到2252cm-1,延伸到967和922cm-1的C-CN带也转移到较低的波数。这可能归因于锡通过-Cequiv;N季铵阳离子(TBMA )、-CH2/氯离子的相互作用溶解TBMACl,这有利于TBMACl的解离。在四甲基氯化铵/乙腈(ACN)和四丁基氯化铵/乙腈的混合物中也报道了类似的溶剂化过程。[49]对于SN-TBMACl-SPE膜中的TBMACl,2875 cm-1处与CH3对称拉伸振动有关的带向8 cm-1的更高波数移动,[50]和1179 cm-1处C-C-C-N的对称拉伸振动峰移向13 cm-1的更高波数。[51] XRD结果证实了TBMACl与SN之间存在较强的化学相互作用。对于二元PEO-TBMACl-SPE薄膜,虽然TBMACl的大部分吸收峰与PEO的吸收峰重叠,但仍能观察到一些明显的变化。分配给2960和1471 cm-1处的CH2拉伸振动的频带向较低的波数移动约4 cm-1。[50] 1471cm-1处CH3的非对称弯曲振动峰值向22cm-1的较高波数移动,峰值强度急剧下降。在二元SPE膜中,800 cm-1处的CCC对称拉伸峰出现在792 cm-1的较低波数处。[52] 与889cm-1处的CH2和CH3摇摆振动相对应的宽峰消失了,[51],取而代之的是,在二元SPE膜中形成了878,898和918cm-1处的新峰,其中归因于10461095和1060cm-1处的PEO的C-O-C拉伸三重态的带宽减小了,[53] 1060cm-1处的峰值显示强度急剧降低。PEO在945cm-1处的CH2对称摇摆峰强度也减弱了。[53]此外,PEO的CH2拉伸在2840-2925cm-1范围内的峰变窄了。[53,54]这些变化证实了二元PEO-TBMACl-SPE膜中PEO的醚基与季铵阳离子的相互作用,有助于TBMACl的分离。对于二元PEO-SN体系,系统研究表明PEO与SN之间的络合是通过SN的CH2与PEO的氧形成氢键[45]来实现的,在与SN的CH2的相互作用中与氯离子竞争。ternay-PEO-TBMACl-SN-SPE薄膜与二元PEO-TBMACl-SPE薄膜具有相似的FTIR图谱。但是,可以注意到一些变化。在2840-2925cm-1处,CH2拉伸峰的宽度进一步减小。1100 cm-1左右的COC拉伸峰和1279 cm-1处的CH2扭转峰进一步减弱。2257cm-1处SN的氰化物峰显著降低,并进一步红移到2248cm-1。因此,TBMACl和SN在PEO矩阵中都得到了很好的协调。SN中的TBMA 离子与非共享电子对和PEO中的醚基之间的溶剂化作用使TBMA 离子对解离,降低了三元SPE的结晶度。此外,SN的CH2更倾向于与PEO中的醚基配位,这不仅进一步降低了PEO的结晶度,而且还导致了氯离子与SN的CH2相互作用的减弱结果表明,氯离子与周围环境的相互作用很弱,预计通过上述相互作用,自由体积增加的PEO链之间氯离子迁移率很高。相反,大的TBMA 离子在PEO链上和链间的迁移受其与聚合物链的显著相互作用的限制。
图2。A,b)光学照片;c,d)SEM图像,e-h)对应的PEO1-TBMACl1-SN3SPE薄膜的元素映射。
用两个不锈钢(SS)封端电极在不同温度下进行阻抗测量,测量离子电导率。支持信息中的图S2显示了不同温度下二元PEO-TBMACl和三元PEO-TBMACl-SN-SPEs的Nyquist图。光谱由离子传导引起的高频凹陷半圆和电极极化引起的低频直线组成。由具有体电阻R的并联R-CPE电路(半圆)和与CPE2(直线)串联的恒相元件CPE1构成的典型等效电路与实测光谱吻合较好。电解质的离子电导率sigma;值可用sigma;=L/(Stimes;R)的公式计算,其中L是膜厚度,S是有效电极面积,如支持信息中的表S4所示。对于所有二元和三元样品,离子电导率随温度呈线性增加。电解质的温度依赖性可以用Arrhenius行为来描述。图4显示了PEO-TBMACl和PEO-TBM
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[246145],资料为PDF文档或Word文档,PDF文档可免费转换为Word
以上是毕业论文外文翻译,课题毕业论文、任务书、文献综述、开题报告、程序设计、图纸设计等资料可联系客服协助查找。
您可能感兴趣的文章
- 纳米材料分散的汽车冷却液配方的腐蚀特性外文翻译资料
- 激光熔化沉积Al-Li合金的组织演变及力学行为外文翻译资料
- 一种高效无载体纳米多孔Co催化剂用于逆水煤气变换反应外文翻译资料
- Zr基块状金属玻璃在NaF和NaCl溶液中的腐蚀机理外文翻译资料
- 一种全固态可充电氯离子电池外文翻译资料
- 纳米氯氧化铁作为可充氯离子电池高性能阴极材料的研究外文翻译资料
- 腐蚀性生物介质中的316L不锈钢与钛合金的比较外文翻译资料
- 三氟化铁作为热电池的高压正极材料外文翻译资料
- 可充电锌空气电池的碱性凝胶聚合物电解质研究外文翻译资料
- 铜/昵钼钮钨(Cu/NbMoTaW)纳米层压板的尺寸依赖性力学 性能和变形机制外文翻译资料

