
英语原文共 17 页,剩余内容已隐藏,支付完成后下载完整资料
通过宏基因组方法探究抗生素的耐药性
摘要:
细菌感染的严重后果已经被抗生素的广泛使用减弱。然而,感染仍是死亡的主要原因,部分原因在于抗药基因的进化和获得。抗生素滥用和开药过量已经成为了一个影响抵抗力选择的驱动力。除了抑制传染性细菌的抗生素耐药性问题,人们很少知道抗性基因的多样性、分布和起源,特别是针对多数不可培养的环境细菌的抗性基因。功能性和基于序列的宏基因组学已经被用于探索新型的抗性决定簇和改进对临床和自然环境中的抗生素耐药机制的理解。本文讨论了通过宏基因组方法来研究抗生素耐药性的最近研究成果和未来的挑战。
正文:
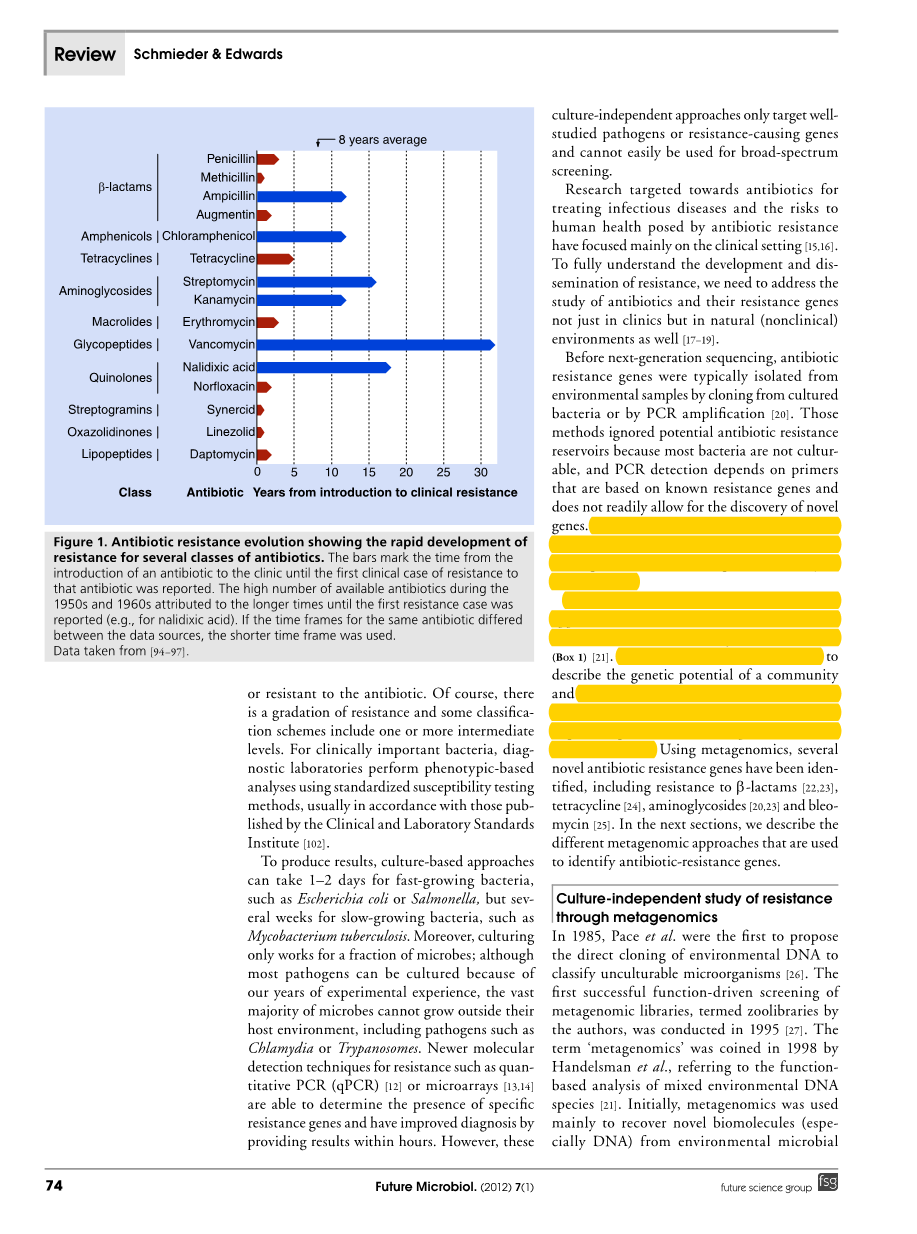
传染性疾病是全球第二大死因和儿童死亡的最重要原因[1,2]。抗生素是目前用于治疗由细菌引起的传染病的最大治疗类别之一,但任何治疗剂的成功使用均受到从首次使用时对该化合物的耐受性或抗性的潜在发展的损害。在临床环境中,病原菌和共生细菌面临着高浓度抗生素的挑战,细菌对大多数发展成熟的抗生素已经产生了耐药性[3]。在医院内,耐药病原体通常在引入新抗生素后几年内出现(图 1)。抗性细菌和抗性基因的传播取决于不同的因素,但主要的原因是抗生素的使用[4]。仅在美国,医院的(与医院有关系的)感染每年会导致大约100,000人死亡,治疗费用超过250亿美元。在全球范围内,估计5-10%进入医院的病人由于逗留而发生感染[101]。尽管在感染性细菌中存在抗生素耐药性的问题,但是人们对于抗性基因的多样性,分布和起源,特别是对于不可培养的大部分环境细菌的抗性基因知之甚少。
抗生素耐药机制
·抗生素的灭活或改变;
·抗生素靶标位点的改变会降低其结合能力;
·代谢途径的改变以规避抗生素的影响;
·降低抗生素的渗透性和/或增加抗生素的主动外排来减少细胞内抗生素的积累.
细菌发展对抗生素的耐药性可通过突变现有基因(垂直进化)[6,7],或通过从其他菌株或物种获得新基因(水平基因转移)来实现[8,9]。通过水平基因转移在细菌之间的基因共享可通过许多不同的机制发生。移动的遗传因素,包括噬菌体、质粒和转座子介导这种转移,在某些情况下,环境中低水平的抗生素的存在是促进基因转移的关键信号[10],可能确保整个微生物群落受到来自抗生素的保护[11]。
抗生素耐药性的检测
抗生素耐药性是一种高度可选择的表型,可以使用在液体培养基中进行的生长抑制剂测定或通过琼脂盘扩散来检测。在基于稀释的生长抑制剂测定中,可以为每种分离的细菌计算抗生素的MIC(最小抑制浓度),这种有机体通常被解释为对抗生素敏感或有抗性。当然,耐药性有等级之分,并且一些分类方案包括一个或多个中间级别。对于临床上重要的细菌,诊断实验室使用标准化的敏感性测试方法进行基于表型的分析,这些方法通常符合临床和实验室标准研究所发布的那些[102]。
为了产生结果,基于培养的方法可以用1-2天获得快速生长细菌,如大肠杆菌或沙门氏菌,但需要数周获得慢速生长细菌,如结核分枝杆菌。此外,培养只适用于一小部分微生物。尽管由于我们多年的实验经验,大多数病原体都可以培养,但绝大多数微生物,包括病原体如衣原体或锥虫,不能在宿主环境外生长。较新的耐药性分子检测技术,如定量PCR(qPCR)[12]或基因芯片[13,14]能够确定特定的抗性基因的存在,并通过在数小时内提供结果来改善诊断。然而,这些免培养的方法仅针对已经有良好研究的病原体或抗性基因,不能轻易地用于广谱筛选。
针对抗生素治疗感染性疾病和抗生素耐药性对人体健康危害的研究主要集中在临床领域[15,16]。要充分了解耐药性的发展和传播,我们不仅要在临床上致力于抗生素及其抗性基因的研究,在自然(非临床)环境中也是如此[17-19]。
在下一代测序之前,抗生素抗性基因通常通过从培养的细菌中克隆或通过PCR扩增从环境样品中分离[20]。这些方法忽略了潜在的抗生素耐药性贮库,因为大多数细菌不能被培养,PCR检测取决于基于已知抗性基因的引物,不容易发现新基因。需要发展免培养技术来鉴定新的抗性基因并获取大多数细菌的遗传多样性。
宏基因组学是克服基于培养或扩增的方法局限性的更现代的方法之一(框1)[21]。这种方法是描述一个群落的遗传潜力、识别群落中微生物的存在类型和负责抗生素耐药性的基因或遗传变异存不存在的有力工具。利用宏基因组学,几种新型抗生素抗性基因已经被鉴定出来,包括对beta;-内酰胺类[22,23],四环素[24],氨基糖苷类[20,23]和博来霉素[25]的耐药性。在接下来的章节中,我们描述了用于鉴定抗生素抗性基因的不同的宏基因组方法。
通过宏基因组学对耐药性的免培养研究
1985年,佩斯等人第一次提出直接克隆环境DNA以分类不可培养微生物[26]。第一次成功的宏基因组文库的功能驱动筛选,被作者命名为zoolibraries,是在1995年进行的[27]。 1998年由汉德尔斯曼等人提出的“宏基因组学”一词,是指对混合环境DNA物种的基于功能的分析[21]。最初,宏基因组学主要用于从环境微生物集合体中回收新的生物分子(特别是DNA)。下一代测序技术的发展导致了替代方法的出现,通过这种方法,样品中的部分DNA一同测序,而不考虑克隆。这种方法有时被称为随机群体基因组学,也被称为宏基因组学。宏基因组测序代表了用于分析复杂微生物群落的rRNA测序的强大替代品[20,28],对环境和临床样品中微生物多样性的研究具有重大影响。如今,宏基因组学领域可以大致分为两种不同的方法:功能性宏基因组学和基于序列的宏基因组学(图3)。
功能性宏基因组学
功能性宏基因组学通过耦合的基于活性的筛选,涉及环境DNA在替代宿主中的克隆和异源表达,以发现可能从其序列中看不明显的基因的功能。通过创建表达克隆的基因组片段的功能性宏基因组文库,和直接选择抗生素抗性,与研究未知序列基因相关的传统挑战得以规避。宏基因组分析揭示了以前未知功能的、仅通过序列无法识别的新型抗生素抗性蛋白。功能性宏基因组学已经被用来鉴定编码使抗生素失活的蛋白质的基因,编码多种药物外排泵的基因和赋予叶酸拮抗剂甲氧苄啶抗性的基因。功能性宏基因组学还被应用于群落的培养的分离株[29]。
基于序列的宏基因组学
基于序列的宏基因组学涉及环境DNA的提取和随机测序,包括未培养细菌的DNA。通常,根据大小分离真核细胞,细菌,病毒和游离DNA(过滤或离心),并从合适的级分中提取总DNA。对DNA样品进行测序,并将该样品假定为整个群落的随机部分。然后将宏基因组序列与已在国内和国际数据库中积累的已知序列(参考序列)进行比较,以鉴定已知导致耐药性的抗性基因和/或突变(图3和图4)。使用广泛的参考抗性基因,可以从单个宏基因组预测对多种抗生素的潜在抗药性。宏基因组序列还代表群落的多样性,包括不能培养的菌株,以及用于研究由于抗生素治疗而导致的群落变化的有价值的信息。
迄今为止,已有超过一千种不同的宏基因组被测序,它们来自各种各样的环境,如土壤、海洋、以及人类的肠道(图5)。此外,基于序列的宏基因组方法也已经分析了一些灭绝物种,如猛犸象[30]和尼安德特人[31]。
过去几年生成的大量的序列数据催生了新一代的分析工具(图4)和序列数据库,如IMG [32],MG-RAST [33],CAMERA [34]和序列读取存档[35]。不幸的是,对于所有原始的宏基因组序列数据,没有一个唯一的权威来源,数据库和数据集之间的数据质量和描述也不尽相同。
微生物群落中的抗生素耐药性
抗生素广泛用于临床中来治疗一系列的疾病,最近的研究旨在了解这些治疗对人类微生物群落的影响。此外,抗生素被用于农业环境,这些做法对人类健康的影响是有争议的。在接下来的章节中,我们讨论了抗生素抗性基因的鉴定及其对人体健康的影响;从环境中分离抗生素抗性基因以及新抗性机制进化的影响;以及抗生素的农业用途如何影响人体健康和抗药性进化。
人类相关微生物中的抗生素耐药性
据估计,在任何给定的人体中,微生物的数量是该人体内细胞数量的十倍,独特基因比我们自己的基因组多 100倍[36]。大多数这些微生物居住在肠道,在肠道群落估计有800-1000种不同的细菌种类[37]。那些微生物对于人类生活至关重要。然而,大约80%的肠道微生物群尚未培养[38]。
近年来,作为国际人类微生物组织联合会的一部分,一些大型项目已经开始调查人类的微生物群落。由NIH发起的人类微生物组计划(HMP)于2008年推出,其任务是在5年内尽可能多地对人类微生物群落进行排序,表征和分析[39,40]。HMP的一部分是通过宏基因组学从五个身体部位进行抽样了解微生物:口腔、鼻腔、皮肤、肠道和(女性)健康供体的阴道。HMP还资助了15个示范项目,为期1年,以研究与人体健康相关的微生物群落,并向其中8个项目提供持续资助。除了HMP之外,人类肠道的宏基因组学(MetaHIT)项目[41]还应用宏基因组学研究欧洲人和亚洲人的肠道。
最近对124位个体进行的MetaHIT宏基因组研究表明,存在共同的核心人类肠道微生物[41],但这个核心可能存在于共享功能基因水平上而不是共有的分类群[42]。微生物生态学中的一个悬而未决的问题是选择是在分类单元的水平还是分类单元的功能水平上进行。除了核心微生物组织,在不同的受试者之间,人类肠道微生物群落组成存在差异。许多内部和外部因素,包括饮食,地域,宿主生理状况,疾病状态和肠道本身特征,促使肠道微生物群落的组成,HMP和相关研究将大大扩展我们对这些关系的理解[36, 43,44]。
人类直接和间接通过农业和清洁或美容产品,将身体暴露于药物中的抗生素中。平均而言,一个美国成年人每年要看两次门诊,其中15.3%的访问量产生了抗生素的处方[45]。在临床访视期间规定的抗生素可能对人类微生物群落产生长期影响。群落层面的研究表明,反复抗生素扰动后,肠道微生物群恢复不完整,而且治疗可能会导致微生物转移到不同但稳定的群落[46]。
在没有选择压力的情况下,抗生素抗性菌株在人类宿主环境中持续存在[4,47]。例如,Sommer等人描绘了两个至少1年没有接受抗生素治疗的,无关的健康个体的功能性抗药性水平特征[48]。使用来自572个细菌分离株的宏基因组DNA和基因组DNA的功能筛选来分析受试者的微生物群落。 对向13种不同抗生素赋予抗性的插入克隆的测序揭示了与已知抗性基因进化上不同的95个独特插入片段。健康个体的共生微生物群落中抗性基因的这种不同的基因库,可能潜在地导致新的抗性致病菌株的出现。也可能表明细菌、新型抗性基因和易于培养的人类病原体之间的横向基因转移存在障碍[49]。我们不知道抗性机制来自哪里,但它们很容易获得。例如,一项关于新生儿的研究表明,出生后3天内,可以在其胎便中发现甲氧西林和氟喹诺酮抗性基因和多药外排泵[50]。在第6天,在婴儿粪便样品中发现替考拉宁抗性基因。在这个特定个体中,第92天的发烧之后,伴随着粪便中beta;-内酰胺酶基因的出现和细菌负荷的减少,尽管当时没有报道抗生素的使用。
类似于人肠道微生物群,口腔微生物群的重要属性是其作为抗生素抗性生物体的储库的能力。口腔细菌可以通过吞咽或通过血液容易地到达其他身体部位,并通过例如咳嗽和接吻传播给其他个体。 因此,抗性口腔细菌有机会快速传播。
Diaz-Torres等人鉴定出一种新的四环素抗性决定簇tet37。 他们从人类口腔的微生物群构建了宏基因组文库[24],3年后,他们应用功能性基因组学来鉴定60位成年人口腔微生物群中编码抗生素抗性的基因[51]。在每个文库中都检测到耐四环素和阿莫西林的克隆,前者是每个文库中最常见的耐药性。 他们没有报告他们的患者的抗生素使用情况,所以不知道对他们的实验产生了怎样的影响。
我们的口腔和粪便微生物群在现有的生
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[28479],资料为PDF文档或Word文档,PDF文档可免费转换为Word
以上是毕业论文外文翻译,课题毕业论文、任务书、文献综述、开题报告、程序设计、图纸设计等资料可联系客服协助查找。
您可能感兴趣的文章
- 中国环境保护投资与经济增长:基于31个省的面板数据的实证研究外文翻译资料
- 评估伊朗北部学校空气污染和环境参数与病态建筑综合征关系外文翻译资料
- 2021年新加坡急救指南外文翻译资料
- 加纳塞孔迪-塔科拉迪大都市高中生对艾滋病毒/艾滋病的认知、态度和做法外文翻译资料
- 毒性基因组学方法筛选氯代阻燃剂三(2‐氯乙基)磷酸盐和三(2‐氯异丙基)磷酸盐的潜在健康影响外文翻译资料
- 磷酸三(2- 氯乙基)酯(TCEP)通过激活 hepg2细胞中的人癌途径基因诱发肝毒性外文翻译资料
- 欧洲的健康素养:欧洲健康素养调查(HLS-EU)的比较结果外文翻译资料
- 使用成人饮食行为问卷评估中国年轻人的食欲特征:因素结构、性别不变性和潜在均值差异,以及与BMI的关联外文翻译资料
- 研究PAFSSB絮凝剂在纸浆和造纸废水中的反渗透预,处理作用外文翻译资料
- 三(1-氯-2-丙基)磷酸暴露于斑马鱼会引起神经发育毒性和异常运动行为外文翻译资料

